- 科技報導
文章專區
2021-01-18人工智慧AlphaFold將破解生物學50年難題?解析蛋白質結構的漫漫長路
469 期
Author 作者
朱利亞/清大生物資訊與結構生物研究所博士,現為英國The Francis Crick Institute博士後。
2020年末尾,以AlphaGo創下聲名的DeepMind公司發表人工智慧「AlphaFold」第二代蛋白質結構預測軟體,參賽挑戰在短時間內預測出蛋白質的結構,在準確度評比(Global Distance Test, GDT)高達92.4,獲得眾所矚目與期待,被認為有機會成為「結構生物學」(Structural biology)的新世代突破。
蛋白質作為生物運轉的重要分子,結構生物學解析其立體功能結構,對於理解生物體運作非常重要。然而,要預測蛋白質結構有理論上的困難,過往主要以「眼見為憑」(Seeing is believing)的方法來進行研究。
回溯到16世紀末,顯微鏡發明以後,人們便開始探索肉眼看不到的微觀世界。1895年,德國科學家倫琴(Wilhelm Röntgen)進行陰極射線管的實驗,偶然發現了X射線,這個發現不但促使醫學的進步,科學家們也開始利用X光去觀察小分子的結構。
解析蛋白質結構的主力
「繞射現象」是光束被一連串規則排列的障礙物散射後,散射後的光經過相長性與相消性干涉,可以得到一個特別的「繞射圖譜」;由於X光的波長接近原子間的鍵長(約10奈米)因此其繞射圖譜就可以用來觀察分子的結構。
1913年,布拉格父子提出了布拉格定律(Bragg’s Law),描述X光繞射角度和晶體原子間距離關係。這個定律也開啟了X光結晶學的大門。利用X光繞射圖譜解析蛋白質結構的方法需要高濃度與純度的蛋白質,得到適合的晶體收集數據。透過電腦運算將蛋白質晶體的繞射圖譜進行傅立葉轉換(Fourier transform),並模擬出三維空間的電子雲密度圖。根據得到的電子雲密度圖,以及蛋白質的胺基酸序列,便可以建立出蛋白質的模型。根據「蛋白質數據資料庫」(Protein Data Bank, PDB)2020年的統計,X光結晶學目前仍為解析蛋白質結構的主要方法(圖一)。
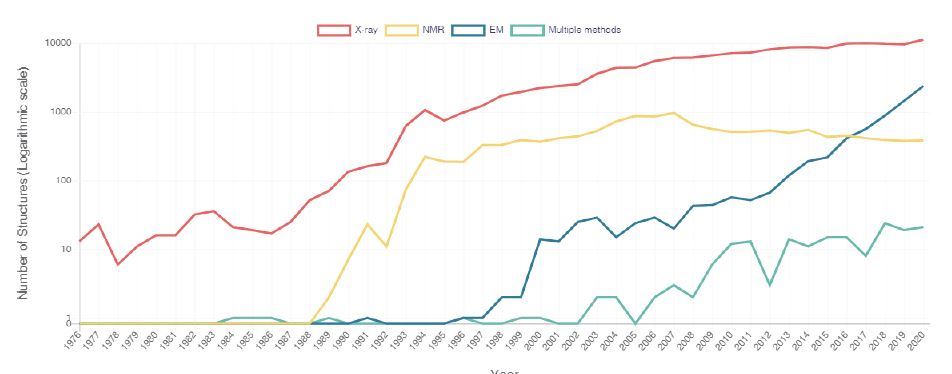 圖一:「蛋白質數據資料庫」(Protein Data Bank, PDB)統計歷年提交的蛋白質結構使用的解析方法。(RCSB PDB, rcsb.org)
圖一:「蛋白質數據資料庫」(Protein Data Bank, PDB)統計歷年提交的蛋白質結構使用的解析方法。(RCSB PDB, rcsb.org)
核磁共振(Nuclear Magnetic Resonance, NMR),是指對固定磁場的原子外加電磁波能量後,原子的自旋方向會翻轉的現象。利用氫原子受到周圍其他原子的影響而改變其吸收能量的頻率,我們可以推測氫原子周圍可能的化學基團並預測出可能的胺基酸。核磁共振技術相較於X光結晶學的優點是不需要將蛋白質進行結晶,可以測得蛋白質在水溶液的狀態下,更接近生理狀態的構型。
NMR雖然可以準確的測得蛋白質在水溶液狀態下的構型,但當蛋白質的分子量增加,NMR圖譜的複雜度也會增加,除了訊號重疊的問題,分析圖譜所需要的時間與人力也相對的增加。因此,NMR主要運用在解析分子量較小的蛋白質結構(< 80 kDa),與X光結晶學一樣為結構生物學主要的技術。
冷凍低溫電子顯微鏡技術突破
2017年的諾貝爾化學獎頒給了冷凍低溫電子顯微鏡(Cryogenic Electron Microscopy, Cryo-EM)。Cryo-EM利用加速電子束打到不規則排列的蛋白質樣品上,電子受蛋白質分子的影響產生散射,散射的電子形成明暗不同的影像,透過電子感測器顯示並記錄影像。再利用電腦將上千或上萬個由此產生的影像重疊,生成高解析度的二維影像,再將二維組合成三維空間的立體影像。
早期的電子顯微鏡受限於電子感測器的靈敏度,因此無法得到高解析度的立體影像。2013年由於電子感測器技術的突破,利用Cryo-EM可以產生超高解析度的立體影像(圖二)。2020年,劍橋大學的團隊利用Cryo-EM得到了超高解析度的原運鐵蛋白(apo-transferrin)結構,其解析度為1.22 Å(Å為長度單位,1 Å
= 10-10m = 0.1 nm) 。
透過Cryo-EM的技術,我們可以解析大分子量的蛋白質以及巨大的蛋白質聚合物的結構,超高的解析度也可以用來觀察蛋白質分子與小分子化合物的結合結構,Cryo-EM也在近年來逐漸成為蛋白質結構解析的主流之一。
經由前面所述的技術,科學家們得以解析出來自於不同物種、不同功能的蛋白質結構。1971年,漢彌爾頓(Walter Hamilton)建立「蛋白質資料庫」(Protein Data Bank,PDB),目標是收錄各實驗室解析出的蛋白質結構,並作為開放平台將資料提供給大眾。2003年,PDB成為了國際性的組織「全球蛋白質資料庫」(Worldwide Protein Data Bank, wwPDB),其成員包含了歐洲(PDBe),美國(RCSB)與日本(PDBj),負責審核、註解與提交世界各國實驗室所提供的蛋白質結構數據。這些實驗數據是蛋白質結構預測、藥物設計,以及蛋白質工程等研究的重要基礎。
AlphaFold預測蛋白質結構最近在《自然》(Nature)期刊上轟動的一則新聞,是關於人工智慧公司DeepMind公司所開發的第二代蛋白質結構預測人工智慧(AI)程式「AlphaFold」,在第14屆的蛋白質結構預測技術的關鍵測試(CASP14)競賽中,從146個團隊中脫穎而出,在準確度評比(Global Distance Test, GDT)中獲得92.4的成績。
GDT主要評測的是預測蛋白質結構的胺基酸中,位置與正確位置差異在固定距離內的胺基酸所佔的比例,比例越高代表預測的結構越準確。GDT分數高於90分的預測結構,已經可以與實驗測得的結構相比擬。這項突破也意味著,將來電腦預測蛋白質結構也許可以和X光繞射、NMR或Cryo-EM成為結構生物學研究的主要工具之一。
蛋白質結構預測起源於1954年,安芬森(Christian B. Anfinsen)發現牛胰核糖核酸酶(bovine pancreatic ribonuclease)在特定環境下,可以攤開成原本的一級結構(primary structure),而當外在環境恢復成原本的狀態時,核糖核酸酶可以重新摺疊成原本具有功能的三級結構。
安芬森根據實驗結果提出安芬森法則(Anfinsen's dogma),即「蛋白質的一級結構(胺基酸序列)可以決定其立體結構」並且「蛋白質的立體構型與其功能有關」。安芬森法則中,出現了從蛋白質序列預測其三級結構的可能性,但由於一條完全展開的多肽鏈(polypeptide chain)有太高的自由度,其可能的構型有太多種,使得蛋白質結構的預測非常困難。
 圖三:安芬森(Christian B. Anfinsen)於實驗室中進行實驗。(By NIH, Public Domain)
圖三:安芬森(Christian B. Anfinsen)於實驗室中進行實驗。(By NIH, Public Domain)
美國科學家利文索爾(Cyrus Levinthal)則於1969年,提出了關於蛋白質折疊的悖論。在他的論文中提到,一個擁有100個胺基酸的多肽鏈中有99個肽鍵(peptide bond),決定每個肽鍵兩側構型的兩面角(dihedral angle)有φ(phi)與ψ(psi)鍵角各一,則總共會產生198個不同的φ與ψ鍵角。假設每一個鍵角在空間中可以有三種構型,則這條多肽鏈在空間中可能摺疊成的構型就會有3198種。假設測試一種可能的折疊方式所需的時間為1皮秒(picosecond,ps),若要測試所有的可能性,則需花費3×1082年。但是,在正常生理條件下,一條多肽鏈折疊成具有功能的蛋白質只需花費幾奈秒(nanosecond, ns),甚至幾皮秒的時間而已。
利文索爾也對這個悖論提出可能的解釋,他認為多肽鏈加速蛋白質折疊的方法,是先由特定區域的胺基酸產生氫鍵等穩定的交互作用,再以這些小區域作為核心,驅動整個三級結構的形成。驅動蛋白質折疊過程的能量,被描述成漏斗狀的能量圖景(funnel-like energy landscape),而蛋白質折疊過程的中間產物,即呈現半折疊狀態的構型也被實驗所證實。
利文索爾也提到,現實中的蛋白質三級結構有可能並非最低能量的折疊狀態,有可能因為要達到最低能量所需的動能不足,使得折疊的狀態停留在能量的局部最小值(local minimum)。目前運用電腦預測蛋白質結構的方法中,有些就是透過分子模擬蛋白質折疊的過程,進而找出蛋白質可能的構型。
 圖四:Google旗下的人工智慧公司DeepMind,本次發表的AlphaFold程式在蛋白質結構預測上獲得了不錯的成績。(DeepMind)
圖四:Google旗下的人工智慧公司DeepMind,本次發表的AlphaFold程式在蛋白質結構預測上獲得了不錯的成績。(DeepMind)
DeepMind公司的程式AlphaFold除了運用上述的結構預測方法,預測局部蛋白質序列的折疊方式,透過分子模擬計算蛋白質折疊的過程,並得到最低能量的結構;還結合了基因序列與蛋白質資料庫的數據,透過序列比對,從基因序列中找出蛋白質的性質,即胺基酸之間的距離與連接胺基酸鍵結的角度(圖五)。AlphaFold訓練神經網路,經由不斷的比對基因序列與蛋白質的性質,持續提高預測蛋白質結構的準確度,最後可以使用整段蛋白質序列進行結構預測,而不需要將蛋白質拆開成不同的片段分開預測。
圖五:DeepMind公司的程式AlphaFold利用基因序列與蛋白質資料庫的數據,透過序列比對從基因序列中找出蛋白質序列中胺基酸之間的距離與連接胺基酸鍵結的角度,訓練神經網路預測準確的蛋白質結構。(The AlphaFold team, DeepMind)
AlphaFold展示了,人工智慧可以整合來自各種不同資料庫的資訊,並從中找出創新並能解決複雜問題的方法。雖然蛋白質結構預測仍無法取代實驗測量的方法,但能夠利用實驗所測得的數據快速且準確的預測結構,並且在未來應用在藥物開發與疾病機制的研究。
期待AI技術跨世代突破,
結構生物學的未來進行式
2019年末爆發的2019冠狀病毒疾病(COVID-19),目前仍沒有可以治療的藥物。而若是能運用AlphaFold這樣的AI技術,預測冠狀病毒組成的蛋白質結構,再針對特定結構如病毒的膜蛋白設計藥物,將可以加速了解這個疾病在分子層面上的致病機制以及解藥的開發。同樣的,AI也可以輔助實驗數據的分析,讓科學家們能夠快速並準確的建立出蛋白質的三維結構模型。雖然AlphaFold在蛋白質結構的預測上,還有許多的進步空間,準確度也尚無法取代實驗技術,但相信不久的將來,AI也可以成為和X光繞射、NMR,以及Cryo-EM等結構生物學技術一樣的跨世代突破。