- 科技報導
文章專區
2018-03-01簡析藥事法修法:新適應症資料專屬權與專利連結專章
435 期
Author 作者
李素華/臺大法律系副教授
2016 年8 月行政院會議通過藥事法修正草案,歷經2017 年5 月25 日立法院社會福利及衛生環境委員會完成審查及爭議條文黨團協商後,同年12 月29 日由立法院三讀通過,2018 年1 月31 日已由總統公布,多數修正條文之施行日由行政院定之。本次修法之最重要內容,乃增訂「新適應症藥品資料專屬權(第40-3 條)」及「西藥之專利連結(第四章之一)」專章,亦即關於藥品智慧財產保護與執行規範。前揭條文,一則在強化新藥智慧財產保護,增加新藥在我國上市的誘因,使病患有使用新藥物及新醫療方式之可能。另一方面,藥事法修法是為轉型發展新藥的國內藥廠提供法律制度面助力,使其能掌握新藥藥廠的專利資訊,以利專利迴避設計及醫藥研發。最後,藥品智慧財產保護之法規健全,亦攸關我國能否與美歐澳日等國建立雙邊貿易關係,「跨太平洋夥伴包容與全面進展協定(Comprehensive and Progressive Agreement for Trans-Pacific Partnership, CPTPP)」區域性貿易協定。
本文以下針對此次藥事法修法議題,先簡述智慧財產保護在醫藥產業所扮演角色與功能、藥品上市程序與智慧財產之關聯性,以瞭解修法的背景緣由,最後再簡析藥事法修法重點。
智慧財產保護對於醫藥研發的重要性
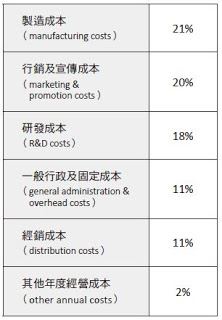
醫藥產業乃高度依賴研發的產業,因科學知識進展使研究人員知悉疾病的成因,亦因新技術及新觀念之持續演進而有新藥品與新醫療方式。藥廠所擁有的研發能力,決定其能否隨著新觀念與新技術而開發新藥物,亦為能否永續經營、持續獲利之關鍵因素。因此,醫藥產業最大的挑戰在於如何導入新產品開發,研發能量成為能否立足於該產業之核心,尤以新藥藥廠為然。歐盟執委會(European Commission)針對醫藥產業之調查結果顯示,2007 年處方藥(prescription medicine)之研發經費占總支出的18%(表一),商業公司統計資料亦呈現相近結果(表二)。相較於其他產業之研發支出通常在4% 以下,顯見研發比重在醫藥產業的重要性。
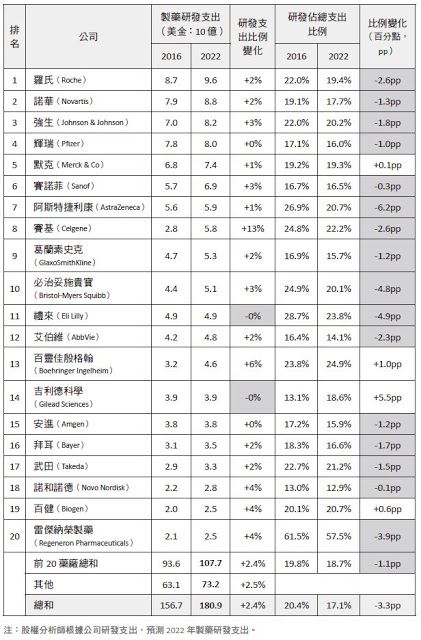
若以個別藥廠所計算的研發投入資料來看(詳見延伸閱讀),2016年羅氏藥廠(Roche Holding AG)之研發經費高達99.15億瑞士法郎(約100.56 億美元),拜耳藥廠(Bayer AG)為46.66 億歐元(約55.12 億美元),BMS 藥廠(Bristol-Myers Squibb Company)為49億美金。我國積極投入研發及轉型發展新藥的藥廠——安成製藥(TWi)亦不遑多讓,101 年度起大幅提高研發支出,104年度的研發費用為7.95億新臺幣,占營收淨額(4.37億新臺幣)的181.71%;105年度的研發費用為7.21 億新臺幣,占營收淨額(7.44 億新臺幣)的96.88%。前揭研發費用雖無法與美歐新藥藥廠商相比,但已顯見醫藥產業之高研發成本現況。另外,若以我國政府投入之科技研發經費相比,105 年度科技部向主計處爭取之科技預算僅有1154 億新臺幣(約38.43 億美元),若合計政府及民間的全國研發經費,亦僅有5414 億元(約180.35 億美元)。與跨國藥廠之經費相互比較後,醫藥產業研發成本之高,更可見一斑。
醫藥產業既然是以研發為基礎,研發成本亦驚人,從事新藥研發的藥廠自然要有雄厚的資金後盾。至於藥廠能否獲得資金奧援的重要關鍵,即在研究成果是否受到智慧財產保護,以確保商業競爭優勢及一定獲利,進而能有足夠的資金持續投入研發,研發所得之新藥品與新醫療方式,亦因而能嘉惠病患。
新藥與學名藥研發成本及上市程序之不同
為了確保藥品之安全性、有效性及品質,從其製造方法、上市及輸入的每一過程,醫藥法規均有嚴密的管理規範,我國除藥事法規定外,另有詳盡及周延的藥品查驗登記審查準則、藥品優良製造、優良運銷規範或準則,即可見一斑。於此法令規範要求下,藥品從一開始的研發、臨床前試驗及臨床試驗階段,歷經層層環節的淘汰程序才能走到藥品上市(圖一),因而造就醫藥產業之研發風險大及時間長。此等特性,同樣回應前述之智慧財產保護重要性。
上述新藥研發流程雖以成分全新的「新成分新藥」為例,但即便是針對已核准藥品的新療效(諸如使用上的新適應症)、新使用途徑(諸如注射改為口服或貼片)、新劑型(諸如膠囊改為錠劑)、新劑量(諸如2 毫升改為10 毫克)、新單位含量(諸如長效劑型或緩釋劑型),均需依循上述研發及試驗流程,證明其用於病患的安全性及療效後,始能准予上市。由此顯見,醫藥法規對於新藥上市的嚴格管理與要求。與新藥研發及試驗流程相對應者,乃各階段所耗費成本。由歐盟資料顯示(表三),新藥研發最主要的成本耗費在三階段的臨床試驗,尤其是第三期臨床試驗。

詳言之,醫藥業者所研發之新產品,要能夠作為藥品上市,必須檢附臨床前及三階段臨床試驗資料,以證明其安全性及療效、能有效用於治療特定疾病,並由醫藥衛生主管機關審核通過。因此,申請新藥上市之藥廠,必須承擔鉅額的新藥研發成本。至於用於治療特定疾病的藥品一旦上市,代表已有足夠的試驗資料證明其安全性與療效。此時,其他藥廠若欲產銷相同成分、相同劑型、相同劑量及相同適應症之藥品,即所謂的「學名藥(generic drug)」,則只需證明該學名藥於人體作用部位的速率(rate)與程度(extent)與對照的新藥相同,亦即具有相同的療效,即可由醫藥衛生主管機關核准上市。
據此,同樣是申請藥品上市,在國內率先推出新藥的藥廠,必須承擔鉅額研發成本,以試驗資料「直接」證明藥品之安全性與療效後才能上市。在新藥藥廠的開疆拓土下,學名藥藥廠能免除重複的臨床前及臨床試驗,藉由引用(refer to)或依賴(rely on)新藥藥廠的試驗資料,以「間接」方式證明學名藥的安全性與療效後,就能立刻上市。兩相比較下,即可得知新藥上市與學名藥上市程序之差異,各該藥廠所需承擔的成本截然不同。
准予藥廠以間接方式證明學名藥符合上市標準,一則是避免資源浪費,免除學名藥藥廠進行重複的安全性及療效試驗。另一方面,正因為學名藥藥廠無需進行上市申請所需試驗,因而能達到學名藥提早上市之目的。
以「資料專屬權」鼓勵藥廠投入新藥研發與新藥上市
為了使藥廠有意願承擔風險及率先投入新藥研發,鼓勵其開疆拓土、進行試驗以取得藥品安全性及療效之上市資料,各國醫藥法規有「資料專屬權」制度。易言之,新藥藥廠耗費鉅額資金所獲得的試驗資料,賦予一定年限的保護,於此期限內,其他藥廠不能以引用或倚賴方式,「間接」使用該等試驗資料而申請學名藥上市;但其他藥廠當然可以自行投入試驗以獲得安全性及療效資料,進而取得藥品上市許可。惟由前述的新藥研發成本結構分析可知,各階段試驗費用過鉅,其他藥廠(尤其是學名藥藥廠)通常並無意願自行進行試驗,而是等到資料專屬權期限屆滿後,再引用新藥資料及以學名藥之姿申請上市。
就制度目的而觀,資料專屬權是對於藥廠投入新藥研發、證明藥品安全性及療效的獎勵;若無藥廠打頭陣向醫藥衛生主管機關申請及核准新藥上市,學名藥藥廠即便有能力製造出相同成分、劑型及劑量的藥品,亦無新藥資料可供引用或倚賴,因而亦無學名藥上市可言。
國際間對於新藥上市申請資料的保護,作法不一,歐盟之資料專屬權長達10 年,亦即第一個新藥上市後,學名藥要等到10年專屬期間屆滿才能上市。日本作法相近,依據新藥種類不同(諸如新成分新藥、新適應症新藥、新劑型新藥或新使用途徑新藥),資料專屬權為4~8 年不等。美、韓及新加坡等國家僅賦予新成分新藥5 年的資料專屬權,其他種類的新藥僅享有3年保護。相較於此,此次修法前我國藥事法第40-2 條第3 項僅有「新成分新藥」之5 年資料專屬權。
近年來國內藥廠積極轉型及從事新藥研發,惟現有能量仍不足以推出新成分新藥,新劑型、新劑量或新適應症新藥仍為主力。但在藥事法規定下,即便能累積臨床試驗資料及就此等新藥申請上市,亦無法享有資料專屬權的獎勵。於此背景下,此次藥事法修正增訂第40-3 條,先就新適應症新藥賦予3 年的資料專屬權;若證明安全性及療效之臨床試驗是在國內執行,則可額外再享有2 年保護(總計5 年)。據此,增訂新適應症新藥資料專屬權之目的,是為了鼓勵我國之醫藥研發、臨床試驗與新適應症新藥上市。醫藥衛生主管機關未來亦應思考,是否仿效美韓新等國家,賦予其他種類新藥3 年資料專屬權,以使國內研發型藥廠能真正獲得獎勵。
以專利連結制度避免侵權爭議及鼓勵新藥研發
除了資料專屬權外,與新藥研發有密切關聯的另一個智慧財產制度為專利權。易言之,資料專屬權所保護的內容,乃新藥上市前的臨床前及臨床試驗資料,在專屬權期間,學名藥藥廠不得引用或依賴;至若專利權所保護標的,則為藥品本身,專利權期間屆至前,學名藥藥廠或任何人,均不得產銷或進口相同藥品。
一、學名藥上市後可能發生專利侵權爭議
前已述及,在現行醫藥法規之規定下,學名藥藥廠申請學名藥上市時,無需自行進行藥品安全性及療效試驗,僅需引用新藥上市申請資料、間接證明學名藥符合上市標準。惟以此方式准予上市的學名藥,無可避免的將發生專利權侵權爭議(圖二)。

准予上市的學名藥若有專利侵權爭議,不僅影響新藥藥廠從事研發的誘因,訴訟中的定暫時狀態處分程序亦可能阻礙學名藥產銷。因此,美、歐、日、加拿大、澳洲及新加坡等國家均有一定的制度,事前避免學名藥進入市場後之專利侵權爭議。歐盟及日本是直接借用資料專屬權制度。以歐盟為例,其賦予新藥長達10 年的資料專屬保護,在10 年的資料專屬權期間,學名藥藥廠不能引用或依賴新藥的試驗資料,在未有資料能證明學名藥的安全性與療效下,自無法取得上市許可。經過長達10 年的資料專屬期間,新藥往往已無專利權保護。期間甚長的資料專屬權固然可避免學名藥與新藥的專利侵權爭議,但長達10 年無法核准學名藥上市,不免使新藥維持較高價格及增加公共衛生支出。相較於此,美國、新加坡及韓國等國家僅賦予新成分新藥5 年及其他新藥3 年之資料專屬權,另以「專利連結制度」來釐清及避免上市學名藥涉訟。
二、藥事法修法草案採行「專利連結」制度避免侵權爭議
我國現行藥事法僅有新成分新藥5 年及新適應症新藥3 年的資料專屬權保護,政策上無意仿效歐盟及日本作法,改採期間甚長的資料專屬權。此次藥事法修法增訂第四章之一「西藥之專利連結」,依循美加新韓等作法,以「專利連結制度」保護新藥研發及避免學名藥侵權。
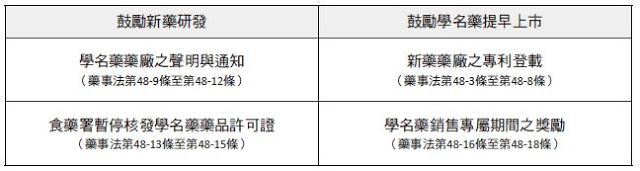
細究此次藥事法修法所增訂的專利連結專章,制度內容看似複雜,實則最核心者僅為如下4 個程序。
(1)新藥藥廠登載及公開專利資訊
在醫藥或任何產業活動,廠商於提供商品或服務前,本應自行檢索智慧局的專利公報或相關資料庫,以瞭解商業行為是否落入他人的專利權範圍、是否有侵權疑慮。但為了減輕學名藥藥廠檢索專利權的負擔及促進學名藥提早上市,專利連結制度之第一個重要程序,是使學名藥藥廠儘速掌握新藥的專利狀態。因此,藥事法新修條文規定,新藥藥廠在取得藥品許可證後,應於法定期間內揭露專利資訊。
( 2)學名藥藥廠透過聲明程序釐清是否侵權
相異於一般技術領域,新藥藥廠既然被額外課以義務,揭露及公告專利權內容及其產品的專利佈局,制度上自應有對應的程序,要求學名藥藥廠先行釐清及確定是否有侵權疑義。若該新藥無任何專利權保護或所有專利權已消滅,醫藥衛生主管機關當可立即准予學名藥上市。反之,該新藥若仍有專利權保護,學名藥藥廠亦無意涉入侵權爭訟,應於所有專利權消滅後才由醫藥衛生主管機關准予學名藥上市。
( 3)12個月內暫停核發藥品許可證以釐清侵權爭議
學名藥上市不會發生侵權爭議的情況,亦即新藥無專利權保護、專利權均已消滅、專利權消滅後學名藥才上市,由醫藥衛生主管機關核准學名藥上市並無疑義。反之,若該新藥仍有專利權保護,藥事法修法條文規定,醫藥衛生主管機關於12 個月內不會准予學名藥上市,以使當事人有一定時間依循智慧局的舉發撤銷或法院的訴訟程序,釐清侵權疑義。為了使學名藥日後能提早上市,於此12 個月內醫藥衛生主管機關不僅繼續審查學名藥申請案,甚而可先核發通過安全性及療效審查的通知函,使學名藥藥廠能先行健保核價,日後能在侵權爭議獲得釐清後立刻上市。
附帶說明者為國際間專利連結制度的暫停核發藥證期間,均參考專利侵權訴訟一審期間與學名藥上市審查期間。美國及新加坡暫停發證期間為30 個月、加拿大24個月、南韓為9 個月,另外中國大陸正研擬建立專利連結制度,目前規劃的期間長達24 個月。若依前述標準來決定暫停發證期間,我國專利連結制度應為15 個月。但為利於學名藥提早上市及考量我國產業需求,藥事法將暫停核發藥證期間縮短為12 個月,等同於學名藥上市申請案的審查及健保核價所需時間。
( 4)首家挑戰專利權的學名藥享有12 個月銷售專屬期間
回應專利連結「促進學名藥提早上市」之立法意旨,學名藥藥廠若積極及勇於挑戰新藥專利權之有效性或從事專利迴避設計,可享有12 個月的銷售專屬期間獎勵,於此12 個月內,醫藥衛生主管機關不會核准其他學名藥上市,確保首家挑戰專利權的學名藥藥廠有一定的利潤。此一制度之目的,是促使學名藥盡快進入市場,達到藥價競爭的結果。
累積智慧財產為生醫產業長遠發展之不可或缺一環
生技醫藥產業高度仰賴創新研發,亦為跨技術領域及提供高附加價值產品與服務的產業。我國自1980 年代開始積極推動生醫產業,陸續核定多項促進產業發展的政策方案,亦有單獨的「生技新藥產業發展條例」立法。2016 年政府部門通過「生醫產業創新推動方案」,希冀進一步提升產業成長動能與國際競爭力。因此,關於鼓勵及提升生醫產業發展上,我國已經建構一定的法制環境,近年來亦有越來越多的醫藥業者不再侷限於國內市場,而是透過併購、產品在美歐國家上市及挑戰國外專利權等方式,積極擴展海外市場。
以創新研發為基礎的醫藥產業,法制環境面上不可或缺的一環乃智慧財產制度,不論是資料專屬權或專利權。我國醫藥業者從單純產銷學名藥,已慢慢轉型研發新藥,不論是在藥品或生物技術專利權的累積上,近年來漸有斬獲,對於藥品專利權保護與權利行使,亦日趨重視。
在政府各部門積極準備跨太平洋夥伴包容與全面進展協定的協商及與其他國家建立經貿合作關係,為國內醫藥產業拓展國際市場之際,此次藥事法修法增訂智慧財產有關條文,或為我國發展生醫產業的重要一哩路。